
Page 79 - Basic _ Clinical Pharmacology ( PDFDrive )
P. 79
CHAPTER 4 Drug Biotransformation 65
Acetaminophen CLINICAL RELEVANCE OF DRUG
METABOLISM
NHCOCH 3
COOH The dose and frequency of administration required to achieve
Glucuronidation Sulfation effective therapeutic blood and tissue levels vary in different
O O
OH UDP OH patients because of individual differences in drug distribution and
O ADP SO 3 H rates of drug metabolism and elimination. These differences are
OH CYP2E1 CYP3A4 determined by genetic factors as well as nongenetic variables, such
OH
HONCOCH 3 as commensal gut microbiota, age, sex, liver size, liver function,
circadian rhythm, body temperature, and nutritional and environ-
NHCOCH 3 NHCOCH 3
mental factors such as concomitant exposure to inducers or inhibi-
tors of drug metabolism. The discussion that follows summarizes
COOH the most important of these variables.
O OH
OH O Reactive Toxic O SO 3 H
Intermediates NCOCH 3 Nontoxic Individual Differences
OH sulfate
OH Individual differences in metabolic rate depend on the nature of the
Nontoxic + drug itself. Thus, within the same population, steady-state plasma
glucuronide
levels may reflect a 30-fold variation in the metabolism of one drug
O and only a twofold variation in the metabolism of another.
GSH-Conjugation + Nucleophillic
+GSH Cell Macromolecules
(Protein-SH)
NHCOCH 3 NHCOCH 3 Genetic Factors
Genetic factors that influence enzyme levels account for some of
these differences, giving rise to “genetic polymorphisms” in drug
SG S-Protein metabolism (see also Chapter 5). The first examples of drugs found
OH OH to be subject to genetic polymorphisms were the muscle relaxant
succinylcholine, the antituberculosis drug isoniazid, and the anti-
coagulant warfarin. A true genetic polymorphism is defined as the
occurrence of a variant allele of a gene at a population frequency of
NHCOCH 3
LIVER CELL DEATH ≥ 1%, resulting in altered expression or functional activity of the
gene product, or both. Well-defined and clinically relevant genetic
polymorphisms in both phase I and phase II drug-metabolizing
SCH 2 CHNHCOCH 3
enzymes exist that result in altered efficacy of drug therapy or
OH COOH
Mercapturic Acid adverse drug reactions (ADRs). The latter frequently necessitate
Conjugate dose adjustment (Table 4–4), a consideration particularly crucial
for drugs with low therapeutic indices.
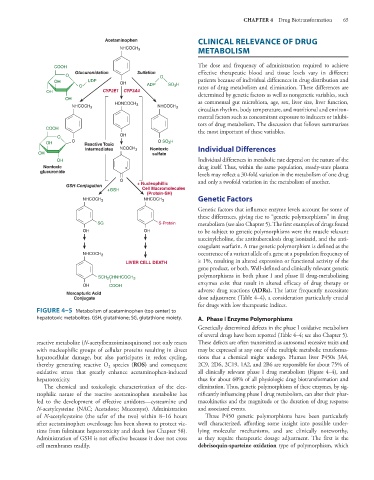
FIGURE 4–5 Metabolism of acetaminophen (top center) to
hepatotoxic metabolites. GSH, glutathione; SG, glutathione moiety. A. Phase I Enzyme Polymorphisms
Genetically determined defects in the phase I oxidative metabolism
of several drugs have been reported (Table 4–4; see also Chapter 5).
reactive metabolite (N-acetylbenzoiminoquinone) not only reacts These defects are often transmitted as autosomal recessive traits and
with nucleophilic groups of cellular proteins resulting in direct may be expressed at any one of the multiple metabolic transforma-
hepatocellular damage, but also participates in redox cycling, tions that a chemical might undergo. Human liver P450s 3A4,
thereby generating reactive O species (ROS) and consequent 2C9, 2D6, 2C19, 1A2, and 2B6 are responsible for about 75% of
2
oxidative stress that greatly enhance acetaminophen-induced all clinically relevant phase I drug metabolism (Figure 4–4), and
hepatotoxicity. thus for about 60% of all physiologic drug biotransformation and
The chemical and toxicologic characterization of the elec- elimination. Thus, genetic polymorphisms of these enzymes, by sig-
trophilic nature of the reactive acetaminophen metabolite has nificantly influencing phase I drug metabolism, can alter their phar-
led to the development of effective antidotes—cysteamine and macokinetics and the magnitude or the duration of drug response
N-acetylcysteine (NAC; Acetadote; Mucomyst). Administration and associated events.
of N-acetylcysteine (the safer of the two) within 8–16 hours Three P450 genetic polymorphisms have been particularly
after acetaminophen overdosage has been shown to protect vic- well characterized, affording some insight into possible under-
tims from fulminant hepatotoxicity and death (see Chapter 58). lying molecular mechanisms, and are clinically noteworthy,
Administration of GSH is not effective because it does not cross as they require therapeutic dosage adjustment. The first is the
cell membranes readily. debrisoquin-sparteine oxidation type of polymorphism, which