
Page 200 - Veterinary Toxicology, Basic and Clinical Principles, 3rd Edition
P. 200
Toxicoproteomics in Diagnostic Toxicology Chapter | 10 167
VetBooks.ir 4700 Reflector Spec #1 MC [BP = 1275.6, 12900]
1222.4373
100 1.2E+4
90
Light-isotope labeled peptide
80 Heavy-isotope labeled peptide
(Control sample)
(Experimental sample)
70
60 1219.4070
% Intensity 50
40
30
20
1217.3799
10 1227.4700 1228.5609 1229.4720 1230.5923 12325217
0
1211.0 1215.4 1219.8 1224.2 1228.6 1233.0
Mass (m/z)
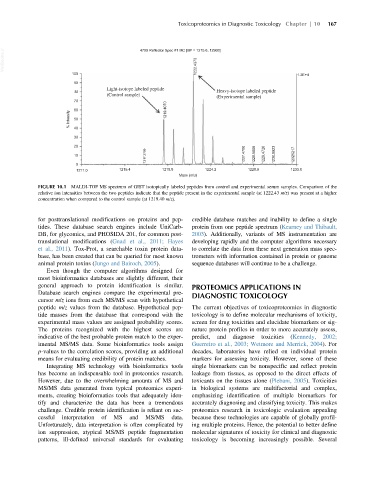
FIGURE 10.1 MALDI-TOF MS spectrum of GIST isotopically labeled peptides from control and experimental serum samples. Comparison of the
relative ion intensities between the two peptides indicate that the peptide present in the experimental sample (at 1222.43 m/z) was present at a higher
concentration when compared to the control sample (at 1219.40 m/z).
for posttranslational modifications on proteins and pep- credible database matches and inability to define a single
tides. These database search engines include UniCarb- protein from one peptide spectrum (Kearney and Thibault,
DB, for glycomics, and PHOSIDA 201, for common post- 2003). Additionally, variants of MS instrumentation are
translational modifications (Gnad et al., 2011; Hayes developing rapidly and the computer algorithms necessary
et al., 2011). Tox-Prot, a searchable toxin protein data- to correlate the data from these next generation mass spec-
base, has been created that can be queried for most known trometers with information contained in protein or genome
animal protein toxins (Jungo and Bairoch, 2005). sequence databases will continue to be a challenge.
Even though the computer algorithms designed for
most bioinformatics databases are slightly different, their
general approach to protein identification is similar. PROTEOMICS APPLICATIONS IN
Database search engines compare the experimental pre- DIAGNOSTIC TOXICOLOGY
cursor m/z ions from each MS/MS scan with hypothetical
peptide m/z values from the database. Hypothetical pep- The current objectives of toxicoproteomics in diagnostic
tide masses from the database that correspond with the toxicology is to define molecular mechanisms of toxicity,
experimental mass values are assigned probability scores. screen for drug toxicities and elucidate biomarkers or sig-
The proteins recognized with the highest scores are nature protein profiles in order to more accurately assess,
indicative of the best probable protein match to the exper- predict, and diagnose toxicities (Kennedy, 2002;
imental MS/MS data. Some bioinformatics tools assign Guerreiro et al., 2003; Wetmore and Merrick, 2004). For
p-values to the correlation scores, providing an additional decades, laboratories have relied on individual protein
means for evaluating credibility of protein matches. markers for assessing toxicity. However, some of these
Integrating MS technology with bioinformatics tools single biomarkers can be nonspecific and reflect protein
has become an indispensable tool in proteomics research. leakage from tissues, as opposed to the direct effects of
However, due to the overwhelming amounts of MS and toxicants on the tissues alone (Plebani, 2005). Toxicities
MS/MS data generated from typical proteomics experi- in biological systems are multifactorial and complex,
ments, creating bioinformatics tools that adequately iden- emphasizing identification of multiple biomarkers for
tify and characterize the data has been a tremendous accurately diagnosing and classifying toxicity. This makes
challenge. Credible protein identification is reliant on suc- proteomics research in toxicologic evaluation appealing
cessful interpretation of MS and MS/MS data. because these technologies are capable of globally profil-
Unfortunately, data interpretation is often complicated by ing multiple proteins. Hence, the potential to better define
ion suppression, atypical MS/MS peptide fragmentation molecular signatures of toxicity for clinical and diagnostic
patterns, ill-defined universal standards for evaluating toxicology is becoming increasingly possible. Several