
Page 93 - Essential Haematology
P. 93
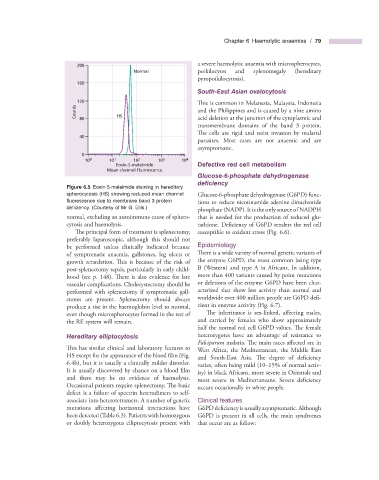
Chapter 6 Haemolytic anaemias / 79
a severe haemolytic anaemia with microspherocytes,
200
Normal poikilocytes and splenomegaly (hereditary
pyropoikilocytosis).
160
South - East Asian o valocytosis
120 This is common in Melanesia, Malaysia, Indonesia
Counts 80 HS and the Philippines and is caused by a nine amino
acid deletion at the junction of the cytoplasmic and
transmembrane domains of the band 3 protein.
The cells are rigid and resist invasion by malarial
40
parasites. Most cases are not anaemic and are
asymptomatic.
0
10 0 10 1 10 2 10 3 10 4
Eosin-5-maleimide Defective r ed c ell m etabolism
Mean channel fluorescence
Glucose - 6 - p hosphate d ehydrogenase
d efi ciency
Figure 6.5 Eosin - 5 - maleimide staining in hereditary
spherocytosis (HS) showing reduced mean channel Glucose - 6 - phosphate dehydrogenase (G6PD) func-
fl uorescence due to membrane band 3 protein tions to reduce nicotinamide adenine dinucleotide
defi ciency. (Courtesy of Mr G. Ellis.)
phosphate (NADP). It is the only source of NADPH
normal, excluding an autoimmune cause of sphero- that is needed for the production of reduced glu-
cytosis and haemolysis. tathione. Deficiency of G6PD renders the red cell
The principal form of treatment is splenectomy, susceptible to oxidant stress (Fig. 6.6 ).
preferably laparoscopic, although this should not
be performed unless clinically indicated because Epidemiology
of symptomatic anaemia, gallstones, leg ulcers or There is a wide variety of normal genetic variants of
growth retardation. This is because of the risk of the enzyme G6PD, the most common being type
post - splenectomy sepsis, particularly in early child- B (Western) and type A in Africans. In addition,
hood (see p. 148) . There is also evidence for late more than 400 variants caused by point mutations
vascular complications. Cholecystectomy should be or deletions of the enzyme G6PD have been char-
performed with splenectomy if symptomatic gall- acterized that show less activity than normal and
stones are present. Splenectomy should always worldwide over 400 million people are G6PD defi -
produce a rise in the haemoglobin level to normal, cient in enzyme activity (Fig. 6.7 ).
even though microspherocytes formed in the rest of The inheritance is sex - linked, aff ecting males,
the RE system will remain. and carried by females who show approximately
half the normal red cell G6PD values. Th e female
Hereditary e lliptocytosis heterozygotes have an advantage of resistance to
Falciparum malaria. The main races affected are in
This has similar clinical and laboratory features to West Africa, the Mediterranean, the Middle East
HS except for the appearance of the blood fi lm (Fig. and South - East Asia. The degree of defi ciency
6.4 b), but it is usually a clinically milder disorder. varies, often being mild (10 – 15% of normal activ-
It is usually discovered by chance on a blood fi lm ity) in black Africans, more severe in Orientals and
and there may be no evidence of haemolysis. most severe in Mediterraneans. Severe defi ciency
Occasional patients require splenectomy. Th e basic occurs occasionally in white people.
defect is a failure of spectrin heterodimers to self -
associate into heterotetramers. A number of genetic Clinical f eatures
mutations affecting horizontal interactions have G6PD defi ciency is usually asymptomatic. Although
been detected (Table 6.3 ). Patients with homozygous G6PD is present in all cells, the main syndromes
or doubly heterozygous elliptocytosis present with that occur are as follow: